Double FAST
Do you have reams of data? Finding the FAST GUI restrictively slow? If so, try using FAST's batch processing system, Double FAST.
Double FAST loads settings saved during a single run through the FAST GUI and applies them to multiple datasets, allowing rapid processing of very large quantities of data. Automation of the data import, segmentation, feature extraction, tracking and division detection stages of the FAST pipeline are supported, although not all stages need to be included during a single batch processing run.
To use Double FAST, you will need to have the Matlab-based version of FAST on your path and so, by implication, a Matlab license. Batch processing for users without a Matlab license is not currently supported.
Installing Double FAST
The .m files required to run Double FAST are included in the main distribution of FAST, available from the download page. Please ensure that you download option 1, the Matlab-based version of FAST.
To run, double FAST requires that both these files and the main FAST files have been added to your Matlab path.
Using Double FAST script
To run Double FAST, you will need to perform two tasks:
- Run FAST once on a single dataset, using the standard GUI. This generates files containing the settings used for each stage of the analysis.
- Set up the DoubleFAST.m script.
If you are not used to editing code directly, don't panic! All you need to do is tell the software where your datasets are, and what stages in the analysis you would like to perform. By leaving these stages in your hands, Double FAST allows you to flexibly adapt your analysis to many different dataset structures and analytical requirements.
Once you have run FAST on your initial dataset, open the DoubleFAST.m script, available within the DoubleFast directory of your FAST installation. At the top of this script are the key variables that you need to define.
The first set of variables allow you to define the locations of your datasets:
mainRoot = 'C:\path\to\root\'; %Main directory in which all of your datasets is located branches = {'Branch_1','Branch_2','Branch_3'}; %Names of subdirectories within main directory in which each separate dataset is stored batchRoots = fullfile(mainRoot,branches);
By default, this is set up assuming a root-branch structure as shown below:
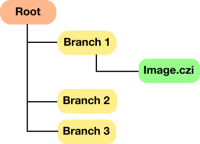
However, this can be flexibly adjusted according to your requirements. The only requirement is that batchRoots is defined as a cell array containing the paths to all of your datasets.
Next, if your datasets are not pre-formatted (i.e. have not been set up according to the format specified by the data importer), you will need to tell FAST the name of your raw datasets:
%Filename of the unprocessed image data (Only need to define this if you are running from raw images) imgName = 'Img.czi';
Note that all of your raw files should have the same name, with their identity specified by their position in the file structure. For example, to process replicate 1 of experimental condition 2, you would give the corresponding raw dataset the filename 'C:\path\to\root\condition_2\replicate_1\Img.czi'.
Finally, you tell Double FAST where it can find the settings files defined during the initial run through FAST:
%The location of the metadata file output during image import (if the bioformats import system is used) MetadataLoc = 'C:\original\analysis\root\Metadata.mat'; %The location of the segmentParams file saved following image segmentation SegmentSettingsLoc = 'C:\original\analysis\root\SegmentationSettings.mat'; %The location of the featSettings file output following feature extraction FeatureSettingsLoc = 'C:\original\analysis\root\CellFeatures.mat'; %The location of the track data generated following the object tracking stages TrackSettingsLoc = 'C:\original\analysis\root\Tracks.mat'; %The location of the track data with divisions detected DivisionSettingsLoc = 'C:\original\analysis\root\Tracks.mat';
If you wish to leave out some part of the main pipeline (for example, because you have already segmented your images using a separate program, or because you don't need to perform division detection), simply comment out the corresponding variables by placing a '%' symbol at the start of the line. To terminate processing at the feature detection stage for example, we would write:
%The location of the metadata file output during image import (if the bioformats import system is used) MetadataLoc = 'C:\original\analysis\root\Metadata.mat'; %The location of the segmentParams file saved following image segmentation SegmentSettingsLoc = 'C:\original\analysis\root\SegmentationSettings.mat'; %The location of the featSettings file output following feature extraction FeatureSettingsLoc = 'C:\original\analysis\root\CellFeatures.mat'; %The location of the track data generated following the object tracking stages %TrackSettingsLoc = 'C:\original\analysis\root\Tracks.mat'; %The location of the track data with divisions detected %DivisionSettingsLoc = 'C:\original\analysis\root\Tracks.mat';
Once you have finished setting up Double FAST, you can run it simply by pressing the 'run' button at the top of the editor, or by pressing F5. Double FAST will now run through each of your datasets one by one, performing the specified operations on each one.